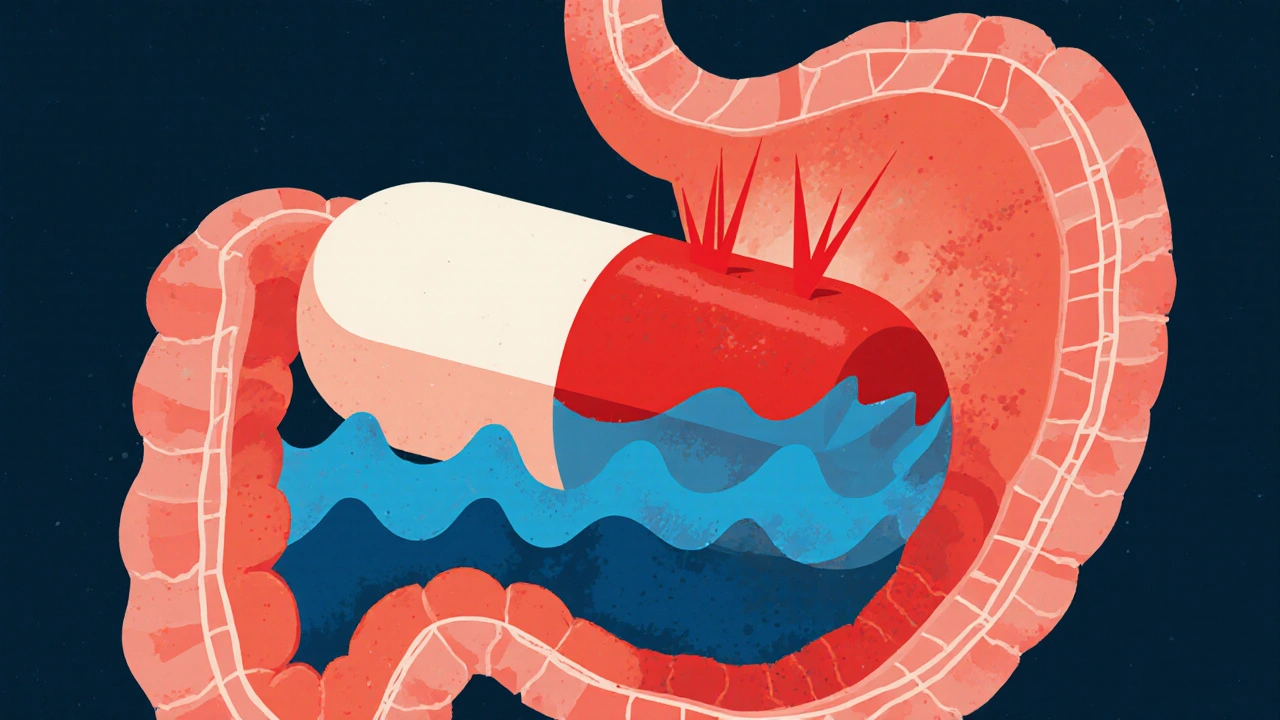
When you take a pill once a day instead of three times, it’s not magic-it’s science. Modified-release (MR) formulations are engineered to control how and when a drug enters your bloodstream. They’re designed to smooth out the peaks and valleys of drug concentration, reducing side effects and improving how well you stick to your treatment. But here’s the catch: just because two pills look the same doesn’t mean they work the same. That’s where bioequivalence comes in-and it’s far more complicated for modified-release drugs than for regular ones.
Why Modified-Release Drugs Are Harder to Match
Immediate-release pills dissolve fast, releasing their full dose in minutes. Bioequivalence for these is straightforward: if the test and reference drugs deliver the same amount of medicine into your blood over the same time, they’re considered equivalent. But with extended-release, delayed-release, or multiphasic formulations, the story changes. These drugs are built with coatings, matrices, or layered beads that control release over hours. A generic version might release 80% of its drug over 12 hours-but if it releases that 80% too fast at first, or too slow later, it could cause side effects or fail to control symptoms.The FDA and EMA don’t just compare total exposure (AUC) and peak concentration (Cmax) anymore. For MR products, they demand detailed profiles. For example, if you’re taking a drug like Ambien CR, which has an immediate-release layer followed by an extended-release layer, regulators need to see that both parts match the brand-name version. That means measuring partial AUC (pAUC) from 0 to 1.5 hours for the quick-release portion, and then again from 1.5 hours to infinity for the slow-release part. Both must fall within the 80-125% range. Miss one, and the application gets rejected.
Regulatory Differences Between Agencies
Not all regulators think the same way. The FDA mostly uses single-dose, fasting studies to test MR generics. Why? Because they believe it’s the most sensitive way to catch differences in how the drug releases from the tablet. The EMA, on the other hand, sometimes still requires multiple-dose studies to reach steady state-especially if the drug builds up in your body over time. This creates headaches for manufacturers trying to get their product approved in both the U.S. and Europe.Take zolpidem (Ambien CR) as an example. The FDA’s 2012 guidance was crystal clear: test both pAUC segments. The EMA didn’t require that. Instead, they focused on half-value duration and midpoint duration time-metrics that measure how long the drug stays in your system, not how it’s released hour by hour. That difference means a generic that passes in Europe might still fail in the U.S. And because most generic manufacturers aim for global approval, they have to design studies that satisfy the strictest standard-which usually means following the FDA’s lead.
Dissolution Testing: The First Line of Defense
Before any human study happens, manufacturers run dissolution tests. These are lab experiments that simulate how a pill breaks down in your stomach and intestines. For extended-release tablets, the FDA requires testing at three pH levels: 1.2 (stomach acid), 4.5 (upper intestine), and 6.8 (lower intestine). Why? Because some coatings dissolve only in certain pH environments. If a generic tablet releases its drug too quickly in acidic conditions, it could cause a spike in blood levels-leading to dizziness or even overdose.Companies often fail at this stage. One formulation scientist at Teva reported that 35-40% of early attempts to match oxycodone ER failed because the dissolution profile didn’t match the brand at pH 6.8. The solution? Switching from standard dissolution apparatus (Apparatus 2) to USP Apparatus 3 or 4, which better mimic how the stomach churns and moves. This isn’t just technical-it’s expensive. A single dissolution method development can cost over $200,000 and take months.

Alcohol and Dose Dumping: A Silent Danger
One of the most dangerous risks with extended-release drugs is alcohol-induced dose dumping. If you take an ER opioid like oxycodone with a glass of wine, the alcohol can dissolve the coating too fast-releasing the entire 24-hour dose in minutes. This isn’t theoretical. Between 2005 and 2015, seven ER products were pulled from the market because of this. The FDA now requires alcohol testing for any ER product containing 250 mg or more of active ingredient. The test involves dissolving the tablet in 40% ethanol and checking how much drug is released in the first hour. If more than 25% of the dose is released, the product fails.Manufacturers now design formulations with alcohol-resistant polymers. But it’s not foolproof. Even small changes in the polymer blend can alter how the tablet behaves with alcohol. That’s why even after a product is approved, the FDA can require post-market studies if new safety concerns arise.
Highly Variable Drugs and RSABE: A Statistical Challenge
Some drugs, like warfarin or clopidogrel, vary wildly in how they’re absorbed from person to person. For these, standard bioequivalence rules (80-125%) don’t work. If the brand drug’s variability is too high, a generic might test as “not equivalent” even if it’s perfectly safe and effective. That’s where Reference-Scaled Average Bioequivalence (RSABE) comes in.RSABE adjusts the acceptance range based on how much the reference drug varies between people. For example, if the reference drug’s within-subject coefficient of variation is over 30%, the FDA allows a wider range-up to 69.84-143.19%. But there’s a cap: the scaling stops at 57.38% standard deviation. This sounds technical, but it’s critical. One Mylan pharmacologist noted that implementing RSABE adds 6-8 months to development time. Why? Because you need more volunteers, more blood draws, and advanced statistical modeling. Fewer than 10% of generic developers have in-house expertise to handle this. Most outsource to specialized CROs, adding over $1 million to the cost.
Why Some Generics Still Fail-Even When They Pass the Tests
Here’s the uncomfortable truth: a generic can meet all regulatory criteria and still cause problems in real life. In 2016, a study in Neurology found that 18% of generic extended-release antiepileptic drugs were linked to higher seizure breakthrough rates than the brand. Patients weren’t failing bioequivalence tests-they were passing them. But the release profile, while statistically equivalent, may have been slightly off in timing. A drug that releases 10% slower over 8 hours might not trigger a failure-but for someone with epilepsy, that delay could mean the difference between control and a seizure.This is why experts like Dr. Donald Mager argue that steady-state studies, while expensive, may be necessary for some drugs. They show how the drug behaves over days, not just hours. The FDA doesn’t require them for most MR products-but for drugs with narrow therapeutic windows, they might be the only way to guarantee safety.

Cost and Complexity: The Hidden Price of MR Generics
Developing a generic immediate-release drug costs about $3-5 million. A modified-release version? $8-12 million. Why the jump? It’s not just the science-it’s the time, the people, and the risk.- Single-dose MR bioequivalence studies cost $1.2-1.8 million, versus $0.8-1.2 million for IR.
- Development timelines stretch by 12-18 months due to complex dissolution testing and statistical analysis.
- Only 3% of MR BE studies are done by small biotechs. The rest are handled by big pharma or contract research organizations (CROs) like Covance and ICON.
And failure rates are high. Between 2018 and 2021, 22% of MR generic applications were rejected for inadequate pAUC data. Another 15% failed because the dissolution profile didn’t match across pH levels. The cost of one failed trial? Up to $1.5 million.
What’s Next? IVIVC, PBPK, and the Future of MR Testing
The future of bioequivalence for MR drugs is moving away from human trials and toward predictive models. In vitro-in vivo correlation (IVIVC) links lab dissolution data directly to how the drug behaves in the body. The FDA has accepted IVIVC for biowaivers in 12 cases, including extended-release paliperidone. If a generic matches the brand’s dissolution profile perfectly, it may not need a human study at all.Even more advanced are physiologically based pharmacokinetic (PBPK) models. These simulate how a drug moves through the body based on anatomy, pH, blood flow, and enzyme activity. Sixty-eight percent of major pharma companies now use PBPK for MR development. It’s still emerging, but it could cut development costs by 40% and reduce the need for human testing.
The FDA plans to release a new guidance in 2024 focused on complex MR products like gastroretentive systems and multiparticulate beads. These are the next frontier-drugs designed to float in the stomach or release in specific parts of the intestine. Testing them will require even more sophisticated tools.
Final Takeaway: Bioequivalence Isn’t Just a Number
Modified-release formulations are one of the most important advances in modern medicine. They make chronic disease management easier, safer, and more convenient. But their complexity means bioequivalence can’t be reduced to a single percentage range. It’s about timing, location, and how the drug behaves in real-world conditions-especially with food, alcohol, or in patients with different gut physiology.For patients, the message is simple: don’t assume all generics are the same. If you’re on an extended-release drug and notice changes in how you feel-side effects, effectiveness, sleep patterns-talk to your pharmacist. For manufacturers, the message is even clearer: mastering MR bioequivalence isn’t just about science. It’s about patience, precision, and understanding that a pill isn’t just a container of medicine. It’s a carefully engineered delivery system.
Why can’t we use the same bioequivalence rules for modified-release and immediate-release drugs?
Immediate-release drugs dissolve quickly and reach peak concentration within hours. Their bioequivalence is measured by total exposure (AUC) and peak level (Cmax). Modified-release drugs are designed to release slowly over many hours, sometimes in multiple phases. A generic might have the same AUC and Cmax as the brand, but if it releases too fast early on or too late later, it can cause side effects or lose effectiveness. That’s why regulators require additional measures like partial AUC and dissolution profiles across different pH levels.
What is dose dumping, and why is it dangerous?
Dose dumping happens when the coating or matrix of an extended-release drug breaks down too quickly, releasing the entire dose at once. Alcohol is a common trigger-it can dissolve the polymer coating meant to control release. For drugs like opioids or stimulants, this can cause overdose, respiratory depression, or heart problems. That’s why the FDA now requires alcohol testing for all ER products containing 250 mg or more of active ingredient.
Are generic modified-release drugs always safe?
Most are. Regulatory agencies require rigorous testing before approval. But bioequivalence doesn’t guarantee identical clinical outcomes in every patient. For drugs with narrow therapeutic windows-like antiepileptics or blood thinners-even small differences in release timing can matter. Studies have shown some generic MR versions have higher rates of breakthrough seizures or INR fluctuations. If you notice changes in how you feel after switching generics, report it to your doctor.
Why do some MR generics cost more than others?
It’s not about the drug-it’s about the development. A simple immediate-release generic costs $3-5 million to develop. A modified-release version can cost $8-12 million because of complex dissolution testing, alcohol studies, statistical modeling (like RSABE), and multiple rounds of trials. If a company fails early, they lose millions. That cost gets passed on in pricing, especially for niche or high-risk MR products.
Can a generic be approved without human studies?
Yes, under specific conditions. If a generic can prove perfect similarity in dissolution profiles across pH levels (f2 ≥ 50) and meets other criteria like drug solubility and permeability, the FDA may grant a biowaiver. This is common for certain extended-release tablets and beaded capsules. In one case, Sandoz got approval for an ER tacrolimus generic using this route, saving $1.5 million and 10 months. But this only works for low-risk, well-understood drugs.
Comments
Rachel Wusowicz November 14, 2025 at 15:49
So... you're telling me the FDA and EMA are basically playing a game of "who can make the most impossible rules" while patients get caught in the middle? And don't even get me started on the "alcohol test"-like, if I have a glass of wine with my pain meds, am I now a criminal? Or is Big Pharma just scared someone might actually enjoy life? I swear, if I find out my pill has a secret polymer coating designed to sabotage my margarita night, I'm filing a class-action lawsuit with glitter and duct tape.
Dan Angles November 15, 2025 at 14:54
The complexity of modified-release bioequivalence underscores a fundamental principle in pharmacology: therapeutic equivalence cannot be reduced to pharmacokinetic metrics alone. The regulatory frameworks established by the FDA and EMA, while divergent in methodology, are both grounded in rigorous scientific inquiry aimed at ensuring patient safety. The use of partial AUC, dissolution profiling across physiological pH gradients, and alcohol challenge studies represents a necessary evolution in regulatory science, reflecting our increasing understanding of drug delivery systems as dynamic, context-sensitive entities.
David Rooksby November 16, 2025 at 13:22
Look, I've been in this game since the 90s and let me tell you-this whole bioequivalence thing is a farce. The FDA wants you to test at pH 1.2, 4.5, and 6.8? Great. But what about when you've got a gut full of beer and pizza? That's pH 3.7, buddy, and no one tests for that. And RSABE? That's just statisticians hiding behind fancy math so they don't have to admit they can't predict how a pill behaves in a 72-year-old with diabetes and three gallstones. I've seen generics pass every test and still make people hallucinate. And the companies? They just tweak the polymer until the machine says "pass," then ship it out. It's not science-it's a rigged casino with a white coat.
Melanie Taylor November 18, 2025 at 06:14
Okay but can we talk about how wild it is that a pill can be "bioequivalent" but still make you feel like a zombie? 😵💫 I switched generics for my epilepsy med and suddenly I was napping at 3pm like it was 2007 again. My pharmacist said "it's the same drug" but... it's not the same *experience*. 🤔 Maybe we need a "feels equivalent" sticker? Like a Yelp review for pills? "4 stars, no dose dumping, but my dreams were weird." 🌙💊
Teresa Smith November 20, 2025 at 05:00
Let me be clear: bioequivalence is not a checkbox. It is a covenant between science and patient well-being. The fact that we still rely on human trials for many modified-release products speaks to the limitations of our predictive models. While IVIVC and PBPK are promising, they are not yet infallible. For drugs with narrow therapeutic windows-antiepileptics, anticoagulants, immunosuppressants-the margin for error is not statistical. It is existential. We must demand more than regulatory compliance. We must demand clinical vigilance. Every patient deserves a drug that behaves as reliably as the one their body learned to trust.
ZAK SCHADER November 20, 2025 at 20:51
USA best. FDA knows what's up. Europe? They let anything through. My cousin took a "generic" Ambien from Germany and woke up driving his car to Canada. No memory. Just a parking ticket and a Starbucks cup. Meanwhile, the FDA makes em test with alcohol? Good. That's what real science looks like. Not some European "midpoint duration" nonsense. We don't play around here. If you can't pass our test, you don't sell here. Period. 🇺🇸
Danish dan iwan Adventure November 21, 2025 at 20:08
RSABE is the only statistically valid approach for high-variability drugs. Failure to implement it reflects regulatory immaturity. Dissolution profiles alone are insufficient. IVIVC requires validation. PBPK requires calibration. Human PK remains gold standard for MR. CROs are overpaid. Biowaivers are acceptable only for low-risk, BCS Class I compounds. Everything else is gambling with patient outcomes.
Ankit Right-hand for this but 2 qty HK 21 November 23, 2025 at 06:16
Who the hell cares about pAUC? You're all just afraid of Indian generics. We make better pills than you. Your FDA is a paper tiger. Our labs in Hyderabad have been reverse-engineering your patents since 2003. You think your "alcohol test" is tough? Try 40°C humidity and no AC. We make pills that work in monsoons and power outages. Your "complex formulations"? We build them in our sleep. Stop acting like you invented medicine. You just own the patents.
Oyejobi Olufemi November 23, 2025 at 19:36
Think about it: every pill you swallow is a tiny betrayal. The manufacturer knows exactly how your body will react-but they don't care. They're not trying to heal you. They're trying to pass a test. They don't care if you feel sluggish, or anxious, or like your brain is made of wet cardboard. They just need that 80-125% window. And the regulators? They're just accountants with lab coats. You think this is science? It's corporate theater. A play where the patient is the audience, the drug is the prop, and the profit is the only script that matters. You're not taking medicine. You're taking a performance.
Daniel Stewart November 24, 2025 at 06:20
It's fascinating how we've moved from the crude pharmacokinetics of the 1980s to these intricate, multi-variable models today. The notion that a drug's behavior can be predicted through dissolution profiles, pH sensitivity, and physiological modeling suggests we're approaching a paradigm shift. Yet, we remain tethered to human trials-not because we lack the tools, but because we lack the collective courage to fully trust them. Perhaps the real barrier isn't scientific, but epistemological: we fear what we cannot see, even when we can model it with astonishing precision.
Latrisha M. November 24, 2025 at 14:26
If you're on an extended-release medication and notice a change in how you feel after switching generics, talk to your pharmacist. They can help you track symptoms and report to the FDA's MedWatch program. Don't assume it's "all in your head." Small differences in release timing can matter. Your experience is valid. Your health matters more than corporate margins.
Jamie Watts November 25, 2025 at 01:39
Man I read this whole thing and all I got was that Big Pharma wants you to pay more for pills that work the same. FDA says one thing EMA says another. Some generic works for you but not your cousin. So what? Just stick with the brand if you can afford it. Otherwise stop complaining. We got cheaper pills. That's the whole point. You want magic? Go to Disney. This is medicine.
John Mwalwala November 25, 2025 at 05:22
Let me drop some jargon on you: the dissolution profile isn't just a curve-it's a fingerprint. The polymer matrix is a controlled-release algorithm written in polyethylene oxide. When you introduce ethanol, you're not just dissolving a coating-you're triggering a phase transition in a semi-interpenetrating polymer network. That's why the FDA requires 40% ethanol testing. It's not about booze-it's about simulating the worst-case scenario of gastrointestinal co-ingestion. If your generic can't survive that, it's not a generic. It's a time bomb. And trust me, your GI tract knows the difference.